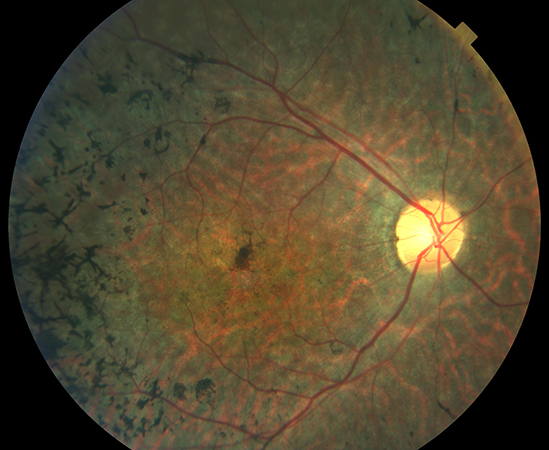
Les malalties minoritàries (també anomenades rares o poc freqüents) es defineixen com aquelles que afecten a 1 o menys de cada 2.000 persones. En conjunt hi ha prop de 10.000 tipus de patologies considerades minoritàries, moltes sense tractament. La recerca és la única estratègia per desenvolupar teràpies per aquestes persones.
Les distròfies hereditàries de la retina (DHR) són un grup heterogeni de malalties rares oculars causades per la mutació d’un sol gen (monogèniques o Mendelianes). Es coneixen més de 300 gens causants de les CHR. Aquestes malalties també es consideren una malaltia genètica de la retina.
No hi ha gaires busquedes per aquest concepte.
A continuació trobareu més informació sobre cadascuna d’aquestes:
Malaltia de Stargardt
La malaltia de Stargardt (OMIM 248200) és una de les distròfies hereditàries de la retina més freqüent. La majoria de casos s’hereten de manera autosòmica recessiva. La disminució de visió que comporta aquesta malaltia afecta habitualment persones joves, amb una incidència sobretot en adolescents i adults joves menors de 20 anys (1:10.000). La malaltia de Stargardt i el fundus flavimaculatus són dos presentacions clíniques de la mateixa malaltia. A nivell histològic, es produeix un cúmul de material tipus lipofuscina a les cèl·lules de l’epiteli pigmentari de la retina per la mutació del gen ABCA4. Aquesta mutació es transmet quan els dos progenitors també posseeixen la malformació d’aquest gen.
Aquesta distròfia hereditàries de la retina provoca una visió desenfocada i sense nitidesa, que dificulta reconèixer rostres i formes, i llegir tant de prop com de lluny i que al final indueix a confondre colors de matisos propers (per exemple, negre i blau marí). També causa dificultat per adaptar-se a la penombra. Encara que no provoca ceguesa absoluta, les persones que la pateixen poden perdre agudesa visual fins a arribar a la ceguesa legal.
Actualment, no existeix cap tractament per aquesta malaltia genetica de la retina, però la recerca en aquest camp continua sent molt activa. Existeixen centres a Europa i Estats Units que estan participant en diferents assajos clínics amb l’objectiu de trobar una teràpia efectiva pels pacients afectats de Stargardt. Aquestes investigacions inclouen tant un apropament farmacològic, com teràpies gèniques i amb cèl·lules mare.
Els principals tractaments farmacològics es divideixen en dos grans grups: els moduladors del cicle visual i els inhibidors del complement. L’Institut de la Màcula, en estreta col·laboració amb la Barcelona Macula Foundation (BMF), és l’únic centre a Espanya que està participant en l’assaig clínic que té per objectiu provar l’eficàcia i seguretat de Zimura® (IVERIC bio; estudi en fase 2b, multicèntric: OPH2005) en pacients amb Stargardt.
Retinosi pigmentària
La retinosi pigmentària (RP) és una de les distrofies hereditàries de retina que provoca una disminució greu de la capacitat visual i que, en molts casos, acaba en ceguesa. Amb una afectació d’1:4.000, a Espanya el nombre d’afectats supera els 15.000 i s’estima que 60.000 persones són portadors dels gens defectuosos i responsables de la malaltia i, per tant, possibles transmissors d’aquesta. L’aparició de la malaltia afecta generalment persones joves i els primers símptomes són la dificultat d’adaptació a la foscor i una pèrdua progressiva del camp visual.
La retinosi pigmentària descriu un grup de malalties hereditàries de la retina que es caracteritzen per una pèrdua progressiva dels fotoreceptors (apoptosi), sobretot els de tipus bastó, i de l’epiteli pigmentari de la retina, degut a mutacions de proteïnes i enzims específics de la mateixa. El component hereditari està present a la meitat dels casos de retinosi pigmentària i el pronòstic, així com la seva progressió, pot estar relacionat amb aquesta herència.
Actualment no es disposa de cap tractament eficaç per a combatre la retinosi pigmentària. La identificació del gen responsable de la malaltia és un aspecte fonamental del maneig clínic del pacient amb RP per la teràpia gènica. Això permet no només la confirmació del diagnòstic, sinó també establir la correlació genotip/fenotip, oferir un pronòstic més acurat i, en el futur proper, indicar un tipus de tractament o un altre.
En l’actualitat, investigadors de la Barcelona Macula Foundation han iniciat el projecte DRUG4SIGHT finançat per La Caixa amb l’IBEC (Institut de Bioenginyeria de Catalunya) i altres entitats d’àmbit estatal per desenvolupar nous fàrmacs en pacients amb degeneracions de la retina. El paradigma d’aquest projecte és completament diferent de les altres aproximacions terapèutiques. Aquí es pretén descobrir i caracteritzar una sèrie de fàrmacs que puguin estimular proteïnes encara presents en la retina degenerada i fer actuar les cèl·lules romanents no degenerades com a fotoreceptors, cèl·lules sensibles a la llum. Els resultats preliminars són prometedors, però caldrà confirmar-los en models animals amb un sistema visual més semblant al de l’ésser humà.
Coroiderèmia
La coroiderèmia (OMIM 300390) és una malaltia recessiva causada per mutacions al gen CHM, localitzat al cromosoma Xq21.2. La mutació provoca la manca d’una proteïna, el Rab escort protein 1 (REP1), que es creu que està involucrada en el tràfic intracel·lular i/o la funció dels bastons. Afecta típicament a homes (1:50.000) i les dones són portadores.
La malaltia provoca una pèrdua de la coriocapilar (la capa de capil·lars més interns de la coroide), l’epiteli pigmentari de la retina i els fotoreceptors (fonamentalment els bastons). S’inicia a la mitja perifèria i progressa de manera centrípeta cap a la màcula, amb uns marges entre retina afectada i sana característicament ben definits. Els símptomes s’inicien habitualment a la joventut. Els pacients noten una pèrdua del camp visual i empitjorament de la visió nocturna que progressen cap a la disminució de la visió central de manera similar a la que s’observa en pacients amb retinosi pigmentària.
Malgrat que no té tractament, la coroiderèmia és una de les malalties hereditàries en les que hi ha més estudis intervencionals registrats (15 a www.clinicaltrials.gov, accedit el 3 de desembre de 2019). Degut a que és una malaltia monogènica, causada per alteracions en un sol gen, el tractament mitjançant teràpia gènica en el que es reemplaça el gen afectat per un que funciona amb normalitat s’està provant amb resultats prometedors. Hi ha assajos a molts països, incloent un assaig clínic en fase III als EUA i Europa, l’estudi STAR.
Amaurosi congènita de Leber
L’amaurosi congènita de Leber (ACL) és una malaltia habitualment autosòmica recessiva present en el moment de néixer o poc després. S’han descrit moltes mutacions en gens diferents com a responsables del seu desenvolupament. S’estima que té una prevalença d’1:40.000 i es pot associar a altres alteracions sistèmiques (retràs del desenvolupament, disfunció renal, etc.).
L’ACL es caracteritza per una degeneració retiniana, en la que un fons d’ull aparentment normal progressa cap alteracions pigmentàries generalitzades, atenuació vascular, atròfia retiniana i del nervi òptic. La visió està severament afectada, i els nadons poden manifestar nistagmus (moviment oscil·latori dels ulls), fotofòbia (sensibilitat anormal a la llum) o estrabisme (mal alineament dels eixos visuals).
En la majoria dels casos no hi ha tractament, encara que es poden adreçar algunes condicions associades a la malaltia (alta hipermetropia, cataractes, etc.). Tot i això, un 10% dels casos d’ACL es deuen a mutacions en el gen RPE65 (ACL tipus 2, OMIM 180069), i en aquests casos es pot utilitzar Luxturna® (voretigene neparvovec, d’Spark Therapeutics), una teràpia gènica basada en el reemplaçament del gen defectuós per un gen de funcional que va mostrar bons resultats visuals. El gener de 2018 va obtenir l’aprovació per part de la FDA (Food and Drug Administration) americana, i a l’abril de 2019 per l’AEMPS (Agencia Española de Medicamentos y Productos Sanitarios). L’aprovació d’aquest fàrmac ha estat una fita, al ser la primera teràpia gènica en oftalmologia aprovada per una agència reguladora.
Retinosquisi lligada a X
La retinosquisi lligada a X (OMIM 312700) és un trastorn ocular bilateral que apareix durant la infància, causat per mutacions en el gen RS1 localitzat al cromosoma Xp22.13. Com el seu nom indica, es transmet lligada al cromosoma X. Afecta només als homes (1/5.000-1/25.000) però les dones en són portadores i tenen un 50% de possibilitats de transmetre la mutació als seus fills.
El gen mutat en la retinosquisi lligada a X (RS1) codifica per la retinosquisina, una proteïna adhesiva que participa en la integritat estructural i funcional de la retina. Així, el fons d’ull es caracteritza per la presència de quists a la zona foveal de la retina i estriacions radials. Els afectats presenten disminució de la visió central i dificultats en la lectura. La pèrdua s’estabilitza durant la joventut per tornar a disminuir cap als 50-60 anys.
Tot i no haver tractament aprovat per la malaltia, avui dia hi ha actius dos assajos clínics (www.clinicaltrials.gov consultat el 3 de desembre de 2019) per avaluar la seguretat, la tolerabilitat i l’eficàcia d’un tractament amb teràpia gènica.
Algunes de les complicacions secundàries a aquesta malaltia són el despreniment de retina (5-22%) i l’hemorràgia vítria (4-40%). És per això que, tot i no haver tractament actualment, es recomana la revisió periòdica per prevenir o tractar de forma immediata qualsevol complicació.
Atròfia Gyrata
L’atròfia Gyrata (OMIM 258870) és una malaltia genetica de la retina per mutacions en el gen que codifica l’ornitina aminotransferasa OAT (10q26). L’activitat deficient d’aquest enzim produeix hiperornitinèmia, però es desconeix encara el mecanisme pel qual condueix a l’atròfia coriorretiniana. És d’herència autosòmica recessiva i la seva prevalença és desconeguda (s’estima 1/50.000). L’edat d’aparició és variable (1 mes-44 anys).
Els primers símptomes són ceguesa nocturna i reducció del camp visual produïts per múltiples àrees circulars d’atròfia coriorretiniana a la perifèria. Amb el pas dels anys, les àrees atròfiques augmenten de mida convergint cap a la màcula i resultant en una pèrdua de la visió central. Els pacients poden presentar associada una miopia amb marcat astigmatisme, cataracta subcapsular posterior d’inici primerenc i edema macular quístic.
S’ha descrit en diversos estudis que una dieta restringida en arginina (precursora de l’ornitina) o una dieta baixa en proteïnes pot reduir el nivell sèric de ornitina, retardant així la progressió de l’atròfia coriorretiniana i de la pèrdua visual. Per aquest motiu és especialment important tenir present aquesta malaltia, ja que la intervenció dietètica pot alentir la seva progressió. Tot i que s’han dut a terme alguns assajos clínics amb teràpia gènica, actualment no n’hi ha cap actiu (www.clinicaltrials.gov consultat el 3 de desembre de 2019).
Fundus Albipuntactus
El fundus albipuntactus (OMIM: 136880) és una de les distrofies hereditàries de retina que debuta a la infància. Aquesta patologia és hereditària, pot ser autosòmica dominant o recessiva i la seva prevalença és desconeguda. La forma autosòmica dominant es produeix per una mutació en el gen PRPH2 i la forma recessiva s’associa a una mutació en el gen RDH5.
El gen RDH5 està involucrat en el cicle visual. Aquest gen és l’encarregat de proporcionar les instruccions a l’enzim 11-cis retinol deshidrogenasa 5, que juga un paper important en la codificació de la llum en senyals elèctriques. Per això, les mutacions d’aquest provocaran un dèficit en la funció que realitza l’enzim 11-cis retinol deshidrogenasa 5, alterant el cicle visual i, en conseqüència, la visió, sobretot en condicions de baixa il·luminació.
A nivell ocular, el fundus albipuntactus es caracteritza per l’aparició de nombroses lesions retinianes petites arrodonides i de color blanc-groguenc que es distribueixen sobretot en mitja perifèria, sense alteració macular.
Els pacients afectats amb aquesta malaltia presenten una ceguesa nocturna no progressiva (amb temps prolongats d’adaptació de llum a foscor), de manera que és important diferenciar-los d’altres entitats progressives amb símptomes semblants, com la retinosi pigmentària. La seva visió en condicions normals d’il·luminació està conservada.
En l’actualitat, als Estats Units s’està portant a terme un estudi que s’encarrega de registrar en una base de dades a pacients amb malalties hereditàries degeneratives de la retina. La creació d’aquestes bases de dades permetrà un major coneixement de la malaltia i la seva història natural, així com aportar dades sobre la seva prevalença.
La distròfia macular vitel·liforme de Best
La distròfia macular vitel·liforme de Best (BVMD) (OMIM 153700) és una de les distrofies hereditàries de retina que debuta a l’edat infantil i a l’adolescència. Aquesta patologia presenta una prevalença d’1 a 9 casos de cada 100.000, afecta més a homes que a dones (3:1) i és hereditària autosòmica dominant.
A la majoria dels casos, la BVMD és causada per una mutació en el gen BEST1 (localitzat en el cromosoma 11q12). Aquest gen és l’encarregat de codificar la bestrofina-1, un canal de clor que s’expressa en l’epiteli pigmentari de la retina, per la qual cosa una alteració en aquesta proteïna provoca l’acumulació de lipofuscina a conseqüència d’un intercanvi iònic anòmal.
La BVMD es caracteritza per una lesió groguenca en la màcula en forma de “rovell d’ou”, degut a l’acumulació anòmala de lipofuscina entre els fotoreceptors i l’epiteli pigmentari de la retina. En estadis més avançats, es produeix una atròfia de l’epiteli pigmentari de la retina . Els pacients que pateixen aquesta malaltia presenten, per norma general, una disminució de l’agudesa visual central, metamorfòpsies, protanopia i disminució de l’índex d’Arden en l’electrooculograma. Per altra banda, la visió perifèrica i l’adaptació a la foscor són normals.
Amb el temps, la BVMD pot evolucionar en una atròfia geogràfica i una de les complicacions associades és l’aparició d’una membrana neovascular subfoveal coroidal, encara que no acostuma a presentar-se en nens.
Actualment, els tractaments que es porten a terme en pacients amb aquestes distrofies hereditàries de retina són per tractar les possibles complicacions derivades de la malaltia, com és l’ús d’anti-VEFG en cas que aparegui una membrana neovascular subfoveal coroidal. Els estudis realitzats en els darrers anys han permès identificar els diferents patrons d’autofluorescència presents en aquesta patologia. Per això, és important fer un seguiment de la malaltia a llarg termini, ja que les modificacions clíniques en l’ autofluorescència permetran un major coneixement de la mateixa i el seu pronòstic en funció del patró. Per altra banda, als Estats Units s’estan realitzant diferents estudis que inclouen la teràpia gènica i amb cèl·lules mare.
Síndrome d’Usher
La síndrome d’Usher (SU) és una de les distrofies hereditàries de retina autosòmica recessiva que es caracteritza per l’associació de la sordesa neurosensorial (generalment congènita) amb la retinosi pigmentària (RP) i la pèrdua progressiva de visió. L’inici generalment ocorre durant la infància i té una prevalença d’1-9:100.000 (a Europa, de 3-4:100.000).
S’han identificat 3 tipus de SU basant-se en les diferències de la funció auditiva i vestibular (la retinosi pigmentària és comú en els tres tipus): (a) tipus 1, en el que la pèrdua d’audició és congènita, profunda i no existeix funció vestibular; la retinosi pigmentària no es manifesta en el naixement i les mutacions impliquen cinc gens (MYO7A, USH1C, CDH23, PCDH15, USH1G) y un locus (USH1E); (b) tipus 2, amb una sordesa congènita menys severa amb funció vestibular preservada, i mutacions en els gens USH2A, GPR98 y DFNB31 i possible locus (15q); i (c) tipus 3, amb inici més tardà tant de la sordesa com de la retinosi pigmentària, amb mutacions en CLRN1.
Tot i les moltes investigacions sobre la síndrome, actualment no hi ha tractament. La recerca es centra en el desenvolupament de teràpies gèniques mitjançant injeccions subretinianes, implants intraoculars d’agents neuroprotectors o els anomenats “ulls biònics”. S’ha de tenir en compte la necessitat de treballar amb un equip multidisciplinar que també inclogui otorrinolaringòlegs, logopedes i psicòlegs. Els implants coclears poden ajudar a mantenir una resta auditiva.
Distròfia de Cons
La distròfia de cons (OMIM 602093) és un grup de malalties hereditàries retinals amb un patró autosòmic recessiu o dominant. Es presenta generalment a la infància o en els primers anys de la vida adulta i la seva prevalença és desconeguda, mentre que en la distròfia conjunta de cons i bastons s’estima que es troba al voltant d’1:40.000 persones.
Els símptomes característics són la pèrdua progressiva d’agudesa visual, visió del color i fotofòbia (sensibilitat anormal a la llum). A l’electrorretinograma estan afectats només els cons, amb preservació de bastons. En alguns casos, en fases avançades poden veure’s afectats els bastons, denominant-se distròfia de cons i bastons; en aquests casos l’electrorretinograma mostra també afectació dels bastons. Oftalmoscòpicament, aquestes distròfies es caracteritzen per la presència de depòsits de pigments en la regió macular, que pot adquirir un aspecte d’“ull de bou”.
Tant la distròfia de cons com la de cons i bastons s’han associat a mutacions en el gen GUCA1A i en els cromosomes 6q, 17p i 19q. Altres malalties on hi ha una afectació selectiva dels cons inclouen l’acromatòpsia o la síndrome d’increment dels cons S. Les alteracions en la visió del color lleus són entitats diferents de la distròfia de cons.
Actualment no existeix un tractament que aturi l’evolució d’aquesta condició i restauri la visió, però hi ha estratègies dirigides a ajudar als pacients a atenuar l’impacte social i psicològic de la pèrdua visual. Les ajudes de baixa visió (sistemes d’augment, filtres) poden millorar la qualitat de vida d’aquests pacients.
Bibliografia
Yannuzi, L. A. (2016) The retina atles. Elsevier
Agarwal, A (2011) Glass’ Atlas of macular diseases. Elsevier
Online Mendelian Inheritance in Man (2019). Disponible en http://www.omim.org.
Clinical Trials (2019). Disponible en http:///www.clinicaltrials.org
El portal sobre enfermedades raras y medicamentos huérfanos (2019). Disponible en http://www.orpha.net
Autors: Clara Abadías, Marc Biarnés, Miriam Garcia i Cristina Romero
Les malalties minoritàries (també anomenades rares o poc freqüents) es defineixen com aquelles que afecten a 1 o menys de cada 2.000 persones. En conjunt hi ha prop de 10.000 tipus de patologies considerades minoritàries, moltes sense tractament. La recerca és la única estratègia per desenvolupar teràpies per aquestes persones.
Les distròfies hereditàries de la retina (DHR) són un grup heterogeni de malalties rares oculars causades per la mutació d’un sol gen (monogèniques o Mendelianes). Es coneixen més de 300 gens causants de les CHR. Aquestes malalties també es consideren una malaltia genètica de la retina.
No hi ha gaires busquedes per aquest concepte.
A continuació trobareu més informació sobre cadascuna d’aquestes:
Malaltia de Stargardt
La malaltia de Stargardt (OMIM 248200) és una de les distròfies hereditàries de la retina més freqüent. La majoria de casos s’hereten de manera autosòmica recessiva. La disminució de visió que comporta aquesta malaltia afecta habitualment persones joves, amb una incidència sobretot en adolescents i adults joves menors de 20 anys (1:10.000). La malaltia de Stargardt i el fundus flavimaculatus són dos presentacions clíniques de la mateixa malaltia. A nivell histològic, es produeix un cúmul de material tipus lipofuscina a les cèl·lules de l’epiteli pigmentari de la retina per la mutació del gen ABCA4. Aquesta mutació es transmet quan els dos progenitors també posseeixen la malformació d’aquest gen.
Aquesta distròfia hereditàries de la retina provoca una visió desenfocada i sense nitidesa, que dificulta reconèixer rostres i formes, i llegir tant de prop com de lluny i que al final indueix a confondre colors de matisos propers (per exemple, negre i blau marí). També causa dificultat per adaptar-se a la penombra. Encara que no provoca ceguesa absoluta, les persones que la pateixen poden perdre agudesa visual fins a arribar a la ceguesa legal.
Actualment, no existeix cap tractament per aquesta malaltia genetica de la retina, però la recerca en aquest camp continua sent molt activa. Existeixen centres a Europa i Estats Units que estan participant en diferents assajos clínics amb l’objectiu de trobar una teràpia efectiva pels pacients afectats de Stargardt. Aquestes investigacions inclouen tant un apropament farmacològic, com teràpies gèniques i amb cèl·lules mare.
Els principals tractaments farmacològics es divideixen en dos grans grups: els moduladors del cicle visual i els inhibidors del complement. L’Institut de la Màcula, en estreta col·laboració amb la Barcelona Macula Foundation (BMF), és l’únic centre a Espanya que està participant en l’assaig clínic que té per objectiu provar l’eficàcia i seguretat de Zimura® (IVERIC bio; estudi en fase 2b, multicèntric: OPH2005) en pacients amb Stargardt.
Retinosi pigmentària
La retinosi pigmentària (RP) és una de les distrofies hereditàries de retina que provoca una disminució greu de la capacitat visual i que, en molts casos, acaba en ceguesa. Amb una afectació d’1:4.000, a Espanya el nombre d’afectats supera els 15.000 i s’estima que 60.000 persones són portadors dels gens defectuosos i responsables de la malaltia i, per tant, possibles transmissors d’aquesta. L’aparició de la malaltia afecta generalment persones joves i els primers símptomes són la dificultat d’adaptació a la foscor i una pèrdua progressiva del camp visual.
La retinosi pigmentària descriu un grup de malalties hereditàries de la retina que es caracteritzen per una pèrdua progressiva dels fotoreceptors (apoptosi), sobretot els de tipus bastó, i de l’epiteli pigmentari de la retina, degut a mutacions de proteïnes i enzims específics de la mateixa. El component hereditari està present a la meitat dels casos de retinosi pigmentària i el pronòstic, així com la seva progressió, pot estar relacionat amb aquesta herència.
Actualment no es disposa de cap tractament eficaç per a combatre la retinosi pigmentària. La identificació del gen responsable de la malaltia és un aspecte fonamental del maneig clínic del pacient amb RP per la teràpia gènica. Això permet no només la confirmació del diagnòstic, sinó també establir la correlació genotip/fenotip, oferir un pronòstic més acurat i, en el futur proper, indicar un tipus de tractament o un altre.
En l’actualitat, investigadors de la Barcelona Macula Foundation han iniciat el projecte DRUG4SIGHT finançat per La Caixa amb l’IBEC (Institut de Bioenginyeria de Catalunya) i altres entitats d’àmbit estatal per desenvolupar nous fàrmacs en pacients amb degeneracions de la retina. El paradigma d’aquest projecte és completament diferent de les altres aproximacions terapèutiques. Aquí es pretén descobrir i caracteritzar una sèrie de fàrmacs que puguin estimular proteïnes encara presents en la retina degenerada i fer actuar les cèl·lules romanents no degenerades com a fotoreceptors, cèl·lules sensibles a la llum. Els resultats preliminars són prometedors, però caldrà confirmar-los en models animals amb un sistema visual més semblant al de l’ésser humà.
Coroiderèmia
La coroiderèmia (OMIM 300390) és una malaltia recessiva causada per mutacions al gen CHM, localitzat al cromosoma Xq21.2. La mutació provoca la manca d’una proteïna, el Rab escort protein 1 (REP1), que es creu que està involucrada en el tràfic intracel·lular i/o la funció dels bastons. Afecta típicament a homes (1:50.000) i les dones són portadores.
La malaltia provoca una pèrdua de la coriocapilar (la capa de capil·lars més interns de la coroide), l’epiteli pigmentari de la retina i els fotoreceptors (fonamentalment els bastons). S’inicia a la mitja perifèria i progressa de manera centrípeta cap a la màcula, amb uns marges entre retina afectada i sana característicament ben definits. Els símptomes s’inicien habitualment a la joventut. Els pacients noten una pèrdua del camp visual i empitjorament de la visió nocturna que progressen cap a la disminució de la visió central de manera similar a la que s’observa en pacients amb retinosi pigmentària.
Malgrat que no té tractament, la coroiderèmia és una de les malalties hereditàries en les que hi ha més estudis intervencionals registrats (15 a www.clinicaltrials.gov, accedit el 3 de desembre de 2019). Degut a que és una malaltia monogènica, causada per alteracions en un sol gen, el tractament mitjançant teràpia gènica en el que es reemplaça el gen afectat per un que funciona amb normalitat s’està provant amb resultats prometedors. Hi ha assajos a molts països, incloent un assaig clínic en fase III als EUA i Europa, l’estudi STAR.
Amaurosi congènita de Leber
L’amaurosi congènita de Leber (ACL) és una malaltia habitualment autosòmica recessiva present en el moment de néixer o poc després. S’han descrit moltes mutacions en gens diferents com a responsables del seu desenvolupament. S’estima que té una prevalença d’1:40.000 i es pot associar a altres alteracions sistèmiques (retràs del desenvolupament, disfunció renal, etc.).
L’ACL es caracteritza per una degeneració retiniana, en la que un fons d’ull aparentment normal progressa cap alteracions pigmentàries generalitzades, atenuació vascular, atròfia retiniana i del nervi òptic. La visió està severament afectada, i els nadons poden manifestar nistagmus (moviment oscil·latori dels ulls), fotofòbia (sensibilitat anormal a la llum) o estrabisme (mal alineament dels eixos visuals).
En la majoria dels casos no hi ha tractament, encara que es poden adreçar algunes condicions associades a la malaltia (alta hipermetropia, cataractes, etc.). Tot i això, un 10% dels casos d’ACL es deuen a mutacions en el gen RPE65 (ACL tipus 2, OMIM 180069), i en aquests casos es pot utilitzar Luxturna® (voretigene neparvovec, d’Spark Therapeutics), una teràpia gènica basada en el reemplaçament del gen defectuós per un gen de funcional que va mostrar bons resultats visuals. El gener de 2018 va obtenir l’aprovació per part de la FDA (Food and Drug Administration) americana, i a l’abril de 2019 per l’AEMPS (Agencia Española de Medicamentos y Productos Sanitarios). L’aprovació d’aquest fàrmac ha estat una fita, al ser la primera teràpia gènica en oftalmologia aprovada per una agència reguladora.
Retinosquisi lligada a X
La retinosquisi lligada a X (OMIM 312700) és un trastorn ocular bilateral que apareix durant la infància, causat per mutacions en el gen RS1 localitzat al cromosoma Xp22.13. Com el seu nom indica, es transmet lligada al cromosoma X. Afecta només als homes (1/5.000-1/25.000) però les dones en són portadores i tenen un 50% de possibilitats de transmetre la mutació als seus fills.
El gen mutat en la retinosquisi lligada a X (RS1) codifica per la retinosquisina, una proteïna adhesiva que participa en la integritat estructural i funcional de la retina. Així, el fons d’ull es caracteritza per la presència de quists a la zona foveal de la retina i estriacions radials. Els afectats presenten disminució de la visió central i dificultats en la lectura. La pèrdua s’estabilitza durant la joventut per tornar a disminuir cap als 50-60 anys.
Tot i no haver tractament aprovat per la malaltia, avui dia hi ha actius dos assajos clínics (www.clinicaltrials.gov consultat el 3 de desembre de 2019) per avaluar la seguretat, la tolerabilitat i l’eficàcia d’un tractament amb teràpia gènica.
Algunes de les complicacions secundàries a aquesta malaltia són el despreniment de retina (5-22%) i l’hemorràgia vítria (4-40%). És per això que, tot i no haver tractament actualment, es recomana la revisió periòdica per prevenir o tractar de forma immediata qualsevol complicació.
Atròfia Gyrata
L’atròfia Gyrata (OMIM 258870) és una malaltia genetica de la retina per mutacions en el gen que codifica l’ornitina aminotransferasa OAT (10q26). L’activitat deficient d’aquest enzim produeix hiperornitinèmia, però es desconeix encara el mecanisme pel qual condueix a l’atròfia coriorretiniana. És d’herència autosòmica recessiva i la seva prevalença és desconeguda (s’estima 1/50.000). L’edat d’aparició és variable (1 mes-44 anys).
Els primers símptomes són ceguesa nocturna i reducció del camp visual produïts per múltiples àrees circulars d’atròfia coriorretiniana a la perifèria. Amb el pas dels anys, les àrees atròfiques augmenten de mida convergint cap a la màcula i resultant en una pèrdua de la visió central. Els pacients poden presentar associada una miopia amb marcat astigmatisme, cataracta subcapsular posterior d’inici primerenc i edema macular quístic.
S’ha descrit en diversos estudis que una dieta restringida en arginina (precursora de l’ornitina) o una dieta baixa en proteïnes pot reduir el nivell sèric de ornitina, retardant així la progressió de l’atròfia coriorretiniana i de la pèrdua visual. Per aquest motiu és especialment important tenir present aquesta malaltia, ja que la intervenció dietètica pot alentir la seva progressió. Tot i que s’han dut a terme alguns assajos clínics amb teràpia gènica, actualment no n’hi ha cap actiu (www.clinicaltrials.gov consultat el 3 de desembre de 2019).
Fundus Albipuntactus
El fundus albipuntactus (OMIM: 136880) és una de les distrofies hereditàries de retina que debuta a la infància. Aquesta patologia és hereditària, pot ser autosòmica dominant o recessiva i la seva prevalença és desconeguda. La forma autosòmica dominant es produeix per una mutació en el gen PRPH2 i la forma recessiva s’associa a una mutació en el gen RDH5.
El gen RDH5 està involucrat en el cicle visual. Aquest gen és l’encarregat de proporcionar les instruccions a l’enzim 11-cis retinol deshidrogenasa 5, que juga un paper important en la codificació de la llum en senyals elèctriques. Per això, les mutacions d’aquest provocaran un dèficit en la funció que realitza l’enzim 11-cis retinol deshidrogenasa 5, alterant el cicle visual i, en conseqüència, la visió, sobretot en condicions de baixa il·luminació.
A nivell ocular, el fundus albipuntactus es caracteritza per l’aparició de nombroses lesions retinianes petites arrodonides i de color blanc-groguenc que es distribueixen sobretot en mitja perifèria, sense alteració macular.
Els pacients afectats amb aquesta malaltia presenten una ceguesa nocturna no progressiva (amb temps prolongats d’adaptació de llum a foscor), de manera que és important diferenciar-los d’altres entitats progressives amb símptomes semblants, com la retinosi pigmentària. La seva visió en condicions normals d’il·luminació està conservada.
En l’actualitat, als Estats Units s’està portant a terme un estudi que s’encarrega de registrar en una base de dades a pacients amb malalties hereditàries degeneratives de la retina. La creació d’aquestes bases de dades permetrà un major coneixement de la malaltia i la seva història natural, així com aportar dades sobre la seva prevalença.
La distròfia macular vitel·liforme de Best
La distròfia macular vitel·liforme de Best (BVMD) (OMIM 153700) és una malaltia que debuta a l’edat infantil i a l’adolescència. Aquesta patologia presenta una prevalença d’1 a 9 casos de cada 100.000, afecta més a homes que a dones (3:1) i és hereditària autosòmica dominant.
A la majoria dels casos, la BVMD és causada per una mutació en el gen BEST1 (localitzat en el cromosoma 11q12). Aquest gen és l’encarregat de codificar la bestrofina-1, un canal de clor que s’expressa en l’epiteli pigmentari de la retina, per la qual cosa una alteració en aquesta proteïna provoca l’acumulació de lipofuscina a conseqüència d’un intercanvi iònic anòmal.
La BVMD es caracteritza per una lesió groguenca en la màcula en forma de “rovell d’ou”, degut a l’acumulació anòmala de lipofuscina entre els fotoreceptors i l’epiteli pigmentari de la retina. En estadis més avançats, es produeix una atròfia de l’epiteli pigmentari de la retina . Els pacients que pateixen aquesta malaltia presenten, per norma general, una disminució de l’agudesa visual central, metamorfòpsies, protanopia i disminució de l’índex d’Arden en l’electrooculograma. Per altra banda, la visió perifèrica i l’adaptació a la foscor són normals.
Amb el temps, la BVMD pot evolucionar en una atròfia geogràfica i una de les complicacions associades és l’aparició d’una membrana neovascular subfoveal coroidal, encara que no acostuma a presentar-se en nens.
Actualment, els tractaments que es porten a terme en pacients amb BVMD són per tractar les possibles complicacions derivades de la malaltia, com és l’ús d’anti-VEFG en cas que aparegui una membrana neovascular subfoveal coroidal. Els estudis realitzats en els darrers anys han permès identificar els diferents patrons d’autofluorescència presents en aquesta patologia. Per això, és important fer un seguiment de la malaltia a llarg termini, ja que les modificacions clíniques en l’ autofluorescència permetran un major coneixement de la mateixa i el seu pronòstic en funció del patró. Per altra banda, als Estats Units s’estan realitzant diferents estudis que inclouen la teràpia gènica i amb cèl·lules mare.
Síndrome d’Usher
La síndrome d’Usher (SU) és una malaltia hereditària autosòmica recessiva que es caracteritza per l’associació de la sordesa neurosensorial (generalment congènita) amb la retinosi pigmentària (RP) i la pèrdua progressiva de visió. L’inici generalment ocorre durant la infància i té una prevalença d’1-9:100.000 (a Europa, de 3-4:100.000).
S’han identificat 3 tipus de SU basant-se en les diferències de la funció auditiva i vestibular (la retinosi pigmentària és comú en els tres tipus): (a) tipus 1, en el que la pèrdua d’audició és congènita, profunda i no existeix funció vestibular; la retinosi pigmentària no es manifesta en el naixement i les mutacions impliquen cinc gens (MYO7A, USH1C, CDH23, PCDH15, USH1G) y un locus (USH1E); (b) tipus 2, amb una sordesa congènita menys severa amb funció vestibular preservada, i mutacions en els gens USH2A, GPR98 y DFNB31 i possible locus (15q); i (c) tipus 3, amb inici més tardà tant de la sordesa com de la retinosi pigmentària, amb mutacions en CLRN1.
Tot i les moltes investigacions sobre la síndrome, actualment no hi ha tractament. La recerca es centra en el desenvolupament de teràpies gèniques mitjançant injeccions subretinianes, implants intraoculars d’agents neuroprotectors o els anomenats “ulls biònics”. S’ha de tenir en compte la necessitat de treballar amb un equip multidisciplinar que també inclogui otorrinolaringòlegs, logopedes i psicòlegs. Els implants coclears poden ajudar a mantenir una resta auditiva.
Distròfia de Cons
La distròfia de cons (OMIM 602093) és un grup de distrofies hereditàries de retina amb un patró autosòmic recessiu o dominant. Es presenta generalment a la infància o en els primers anys de la vida adulta i la seva prevalença és desconeguda, mentre que en la distròfia conjunta de cons i bastons s’estima que es troba al voltant d’1:40.000 persones.
Els símptomes característics són la pèrdua progressiva d’agudesa visual, visió del color i fotofòbia (sensibilitat anormal a la llum). A l’electrorretinograma estan afectats només els cons, amb preservació de bastons. En alguns casos, en fases avançades poden veure’s afectats els bastons, denominant-se distròfia de cons i bastons; en aquests casos l’electrorretinograma mostra també afectació dels bastons. Oftalmoscòpicament, aquestes distròfies es caracteritzen per la presència de depòsits de pigments en la regió macular, que pot adquirir un aspecte d’“ull de bou”.
Tant la distròfia de cons com la de cons i bastons s’han associat a mutacions en el gen GUCA1A i en els cromosomes 6q, 17p i 19q. Altres malalties on hi ha una afectació selectiva dels cons inclouen l’acromatòpsia o la síndrome d’increment dels cons S. Les alteracions en la visió del color lleus són entitats diferents de la distròfia de cons.
Actualment no existeix un tractament que aturi l’evolució d’aquesta condició i restauri la visió, però hi ha estratègies dirigides a ajudar als pacients a atenuar l’impacte social i psicològic de la pèrdua visual. Les ajudes de baixa visió (sistemes d’augment, filtres) poden millorar la qualitat de vida d’aquests pacients.
Bibliografia
Yannuzi, L. A. (2016) The retina atles. Elsevier
Agarwal, A (2011) Glass’ Atlas of macular diseases. Elsevier
Online Mendelian Inheritance in Man (2019). Disponible en http://www.omim.org.
Clinical Trials (2019). Disponible en http:///www.clinicaltrials.org
El portal sobre enfermedades raras y medicamentos huérfanos (2019). Disponible en http://www.orpha.net
Autors: Clara Abadías, Marc Biarnés, Miriam Garcia i Cristina Romero